Authors:
Abstract
Introduction
Biomaterials can provide localized reservoirs for controlled release of therapeutic biomolecules and drugs for applications in tissue engineering and regenerative medicine. As carriers of gene-based therapies, biomaterial scaffolds can improve efficiency and delivery-site localization of transgene expression. Controlled delivery of gene therapy vectors from scaffolds requires cell-scale macropores to facilitate rapid host cell infiltration. Recently, advanced methods have been developed to form injectable scaffolds containing cell-scale macropores. However, relative efficacy of in vivo gene delivery from scaffolds formulated using these general approaches has not been previously investigated. Using two of these methods, we fabricated scaffolds based on hyaluronic acid (HA) and compared how their unique, macroporous architectures affected their respective abilities to deliver transgenes via lentiviral vectors in vivo.
Methods
Three types of scaffolds—nanoporous HA hydrogels (NP-HA), annealed HA microparticles (HA-MP) and nanoporous HA hydrogels containing protease-degradable poly(ethylene glycol) (PEG) microparticles as sacrificial porogens (PEG-MP)—were loaded with lentiviral particles encoding reporter transgenes and injected into mouse mammary fat. Scaffolds were evaluated for their ability to induce rapid infiltration of host cells and subsequent transgene expression.
Results
Cell densities in scaffolds, distances into which cells penetrated scaffolds, and transgene expression levels significantly increased with delivery from HA-MP, compared to NP-HA and PEG-MP, scaffolds. Nearly 8-fold greater cell densities and up to 16-fold greater transgene expression levels were found in HA-MP, over NP-HA, scaffolds. Cell profiling revealed that within HA-MP scaffolds, macrophages (F4/80+), fibroblasts (ERTR7+) and endothelial cells (CD31+) were each present and expressed delivered transgene.
Conclusions
Results demonstrate that injectable scaffolds containing cell-scale macropores in an open, interconnected architecture support rapid host cell infiltration to improve efficiency of biomaterial-mediated gene delivery.
Keywords
Gene therapy Tissue engineering Injectable scaffold Lentivirus
Abbreviations
- HA
-
Hyaluronic acid
- HA-SH
-
Thiolated hyaluronic acid
- PEG
-
Poly(ethylene glycol)
- FLuc
-
Firefly luciferase
- FLuc-LV
-
Firefly luciferase encoding lentivirus
- PEG-VS
-
Vinyl sulfone-terminated poly(ethylene glycol)
- PEG-mal
-
Maleimide-terminated poly(ethylene glycol)
- DTT
-
Dithiothreitol
Introduction
The physiological regenerative response to injury can be insufficient, resulting in incomplete regeneration and potential re-injury, infection or lower quality of life. This is especially true in cases where secondary conditions affecting wound healing, such as diabetes, are present. Engineered biomaterial scaffolds are being developed to accelerate or enhance tissue regeneration after injury. Injectable, in situ forming scaffolds are ideal for minimally invasive treatment of conditions where the area of injury is difficult to access or ill-defined in structure. In situ-forming scaffolds can facilitate regeneration by providing structural support and tissue-appropriate mechanical properties.8,20,25,57 These properties can be further modulated to support localized delivery of therapeutic drugs, genes and even live cells. Recent reports have found that inclusion of cell-scale macropores into biomaterial scaffolds can support delivery of therapeutic transgenes by accelerating host cell infiltration and nutrient transport throughout implanted scaffolds.43,50
While several techniques have been established to create non-injectable scaffolds with cell-scale macroporosities to improve integration with host tissues or facilitate gene delivery, including electrospinning, gas foaming, salt or particle leaching, freeze-thaw, and phase separation,9,35,36,38,39,41 only more recently have biomaterial scaffolds been developed that are both macroporous and injectable, a capability which enables their minimally invasive delivery to irregularly shaped defects and thus increases their potential for clinical translation. Macroporous, injectable scaffolds have been developed using in situ degradation of sacrificial porogens,16,22,43,47 annealed microparticles,6,18,42,45,52,53 shape-memory polymers4,54 and crosslinked micro-ribbons.19 Such scaffolds can form interconnected networks of macropores with tunable sizes and have shown great promise in improving infiltration of host cells and blood vessels after implantation in vivo.18,43,58 Additionally, macroporous scaffolds have been shown to improve the delivery of therapeutic genes in vivo, primarily by supporting early cell infiltration before vector degradation occurs.43,50 Delivery of therapeutic genes from biomaterial scaffolds can be used to enhance tissue regeneration by modulating expression of factors relating to angiogenesis, stem cell differentiation, inflammation, and numerous other biological processes.17,26,29,32, 33, 34 However, genetic vectors, such as lentivirus, are susceptible to degeneration in vivo, with half-lives as low as 12 h.21,48 Thus, injectable, macroporous scaffolds are attractive vehicles for localized delivery of therapeutic transgenes.
While injectable scaffolds utilizing either sacrificial porogens43 or annealed particles52,53 have been explored previously for their potential as vehicles for gene delivery, the relative potentials of these methods for transgene delivery in vivo remain uncharacterized. Here, we directly compare the abilities of injectable, macroporous scaffolds fabricated using these two methods from composites of polyethylene glycol (PEG) and hyaluronic acid (HA), and their nanoporous hydrogel counterparts, to deliver lentiviral vectors to the mammary fat in mice. PEG is a widely studied bio-inert, synthetic, polymer used commonly for drug delivery and a number of other clinical applications. HA is a linear polysaccharide that is ubiquitous in the extracellular matrix of mammals and highly biocompatible. HA biomaterials are currently being used for several clinical applications, including as dermal fillers, in ophthalmologic applications and for joint viscosupplementation. HA biomaterials can provide several advantages for tissue regeneration and wound repair, including increased angiogenesis and, in its high molecular weight form, reduction of inflammatory response.31
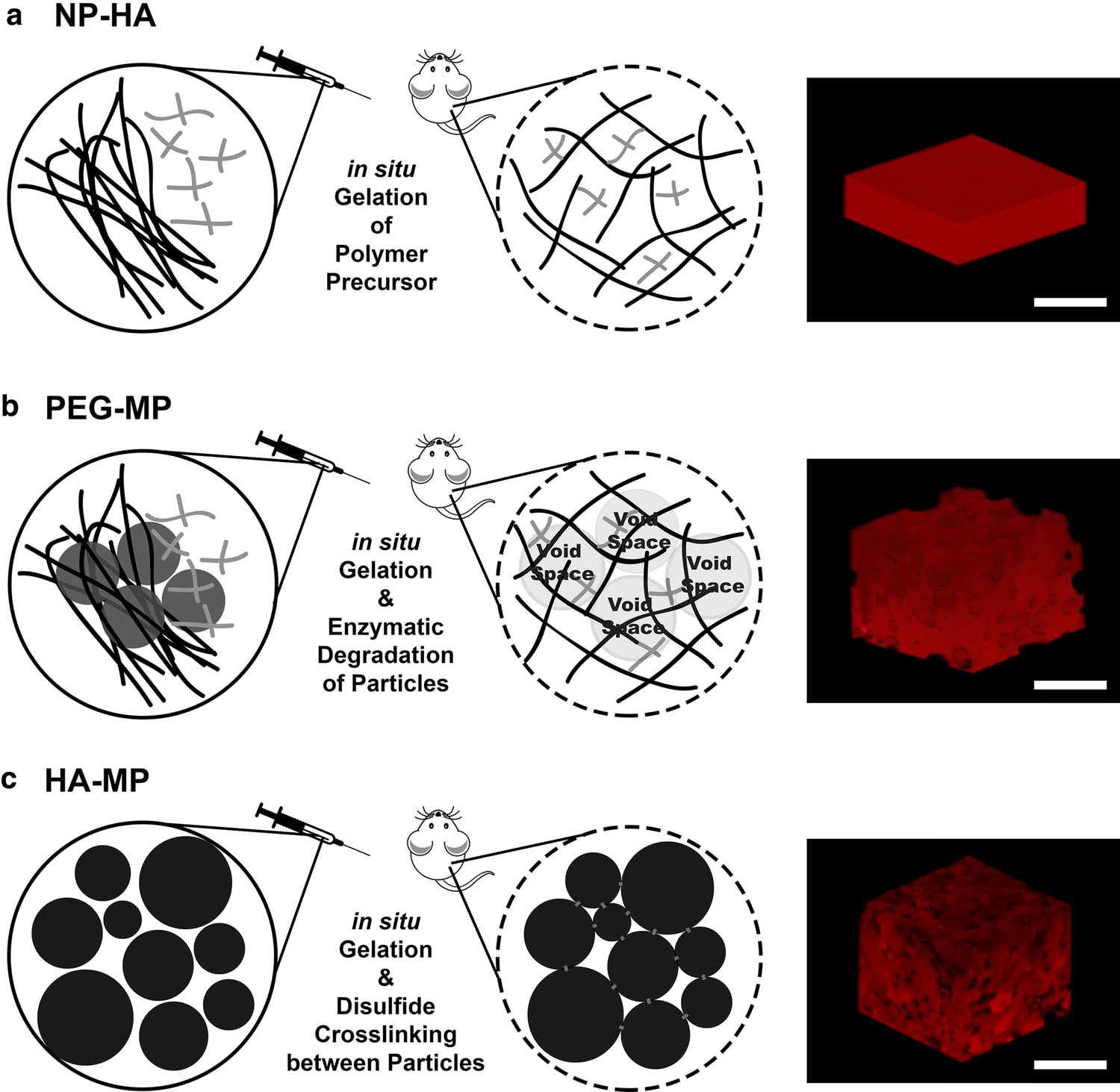
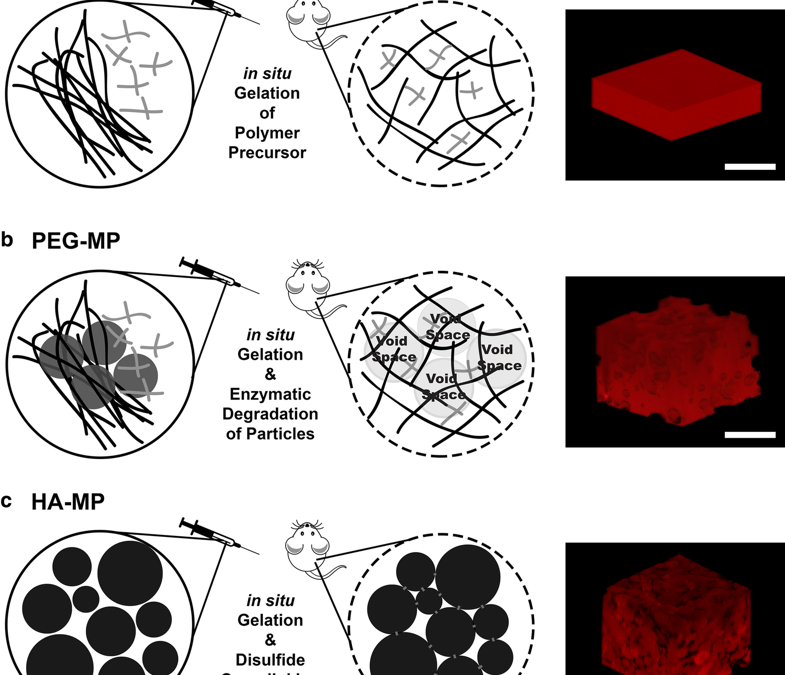
Fabrication schemes for injectable, hyaluronic acid (HA)-based scaffolds with varying porous architectures. NP-HA scaffolds are formed by crosslinking thiolated HA and polyethylene glycol-vinyl sulfone (PEG-VS) into homogenous, nanoporous scaffolds (a). For PEG-MP scaffolds, a nanoporous HA-PEG hydrogel is crosslinked around PEG microparticles that can proteolytically degrade to leave behind a macroporous architecture (b). Covalently crosslinked HA-PEG microparticles can assemble in situ via disulfide bonds to form HA-MP scaffolds with macroscale pores in the void spaces between microparticles (c). RGD peptides are included in all scaffold iterations to support cell adhesion. Scale bars = 500 µm.
Results demonstrate that lentivirus-loaded HA-MP scaffolds improve in vivo transgene expression significantly compared to NP-HA scaffolds. This improvement was paired with increased cell infiltration compared to other scaffolds, particularly of fibroblasts and endothelial cells. However, PEG-MP scaffolds only showed improvement over NP-HA in transgene expression in a few specific cases and no statistically significant improvements in cell infiltration. This report demonstrates the enormous potential of injectable, macroporous scaffolds—formed from in situ-annealing of hydrogel microparticles—to improve biomaterial-mediated gene delivery for a wide range of applications in tissue engineering and regenerative medicine.
Materials and Methods
Materials were purchased from Thermo Fisher Scientific unless otherwise noted.
Synthesis and characterization of thiolated hyaluronic acid (HA-SH)
Sodium hyaluronate (Mw = 700 kDa, LifeCore Biomedical) was dissolved at 10 mg/mL in distilled, deionized (di) H2O and thiolated as previously shown.56 The pH of the HA solution was adjusted to 5.5 using 0.1M HCl. 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide (EDC) was dissolved in di H2O at the appropriate molar ratio (0.25 × unless otherwise stated) immediately before addition to HA solution. Molar ratios in all cases are reported with respect to carboxyl groups on glucuronic acid moieties of HA. N-hydroxysuccinimide (NHS, Acros Organics) was then added at half of the molar ratio as EDC. The pH was then readjusted to 5.5 and the reaction was mixed at room temperature for 45 min. Then, cystamine dihydrochloride (Sigma-Aldrich) was added (0.25 × molar ratio), pH was adjusted to 6.25 using 0.1 M NaOH, and the reaction continued while stirring at room temperature overnight. Dithiothreitol (DTT, Sigma-Aldrich) was added in excess (4 × greater than cystamine) at pH 8. The mixture was stirred for 1–2 h to cleave cystamine disulfides and yield thiolated HA (HA-SH). The reaction was quenched by adjusting the pH to 4. HA-SH was purified using dialysis against acidic (pH 4) di H2O for 3 days in the dark. Purified, HA-SH was filtered through a 0.22 µm filter (EMD Millipore), frozen under liquid nitrogen, lyophilized, and stored at − 20 °C until use. HA thiolation was confirmed using proton NMR spectroscopy and the colorimetric Ellman’s test for free thiols.12
Lentivirus Production
Firefly luciferase (FLuc)-encoding lentivirus (FLuc-LV) and td-tomato-encoding lentivirus, each with a constitutively active CMV promoter, were generated using a third generation packaging system, as previously described.11 Plasmids were generously provided by Prof. Lonnie Shea at the University of Michigan. Briefly, 80% confluent human embryonic kidney cells (Lenti-X 293T Takara Bio USA) were transfected with third generation packaging plasmids (pRSV-Rev and pMDLg/pRRE were gifts from Didier Trono (Addgene plasmids # 12253; #12251), a plasmid encoding the lentiviral capsule (pCMV-VSV-G was a gift from Bob Weinberg, Addgene plasmid # 8454). and FLuc or td-tomato plasmids using jetPRIME transfection reagent (Polyplus transfection). Lentiviral particles were recovered from media after 2 days of culture using PEG-it virus precipitation solution (SBI System Biosciences), resuspended in PBS and stored at – 80 °C. Lentiviral titers were calculated using the Lenti-X qRT-PCR titration kit (Takara Bio USA).
Formation of HA-PEG Scaffolds (NP-HA)
HA-SH and 4-arm vinyl sulfone-terminated PEG (PEG-VS) (20kDa, Laysan Bio) were crosslinked via Michael-type addition between thiol and vinyl sulfone functional groups.24 HA-SH and PEG-VS were dissolved separately in PBS at pH 7.4. Cysteine-terminated RGD peptide (GCGYGRGDSPG, GenScript Biotech) was conjugated PEG-VS by reaction at room temperature for 1 h prior to gel formation to provide sites for cell adhesion.5 For swelling and mechanical characterization studies, l-cysteine (Sigma Aldrich) was used in place of the RGD peptide prior to hydrogel formation. The pH of HA-SH was adjusted to 7.0 using 1 M NaOH after dissolution. For in vitro studies, HA-SH and RGD- or l-cysteine-modified PEG-VS solutions were mixed and pipetted into circular wells of a silicon isolator (8 mm diameter, 1 mm depth, Grace BioLabs)15 with final concentrations of 10 mg/mL of HA-SH, 150 µM of peptide, and 5.93 mg/mL of PEG-VS. Scaffolds were incubated at 37 °C for 2 h to ensure crosslinking had completed.24
Formation of Microparticles for HA-MP and PEG-MP Scaffolds
For HA-MP microparticles, HA-SH was crosslinked using 4-arm, maleimide-terminated PEG (PEG-mal) (20kDa, Laysan Bio). Crosslinking via Michael-type addition of maleimide and thiol groups occurs rapidly near neutral pH, allowing microparticles to quickly crosslink and stabilize within oil emulsions.59 HA-SH and PEG-mal were dissolved separately in PBS adjusted to pH 6.5 using 1M NaOH. Hydrogel precursor solution consisted of PEG-mal mixed with 20 mg/mL HA-SH to yield a mixture with 1.2:1 ratio of thiol:maleimide. For PEG-MP microparticles, PEG-mal (30 mg/mL) and a fast-degrading, plasmin-degradable peptide crosslinker (3.24 mg/mL, Ac-GCYK↓NRGCYK↓NRCG, GenScript)44 were dissolved in PBS at pH 6.5. For both types of microparticles, 100 μL of precursor solution was vortexed in 900 μL mineral oil with 1% span 80 surfactant for 20 s before addition of 100 μL mineral oil containing 0.1% triethylamine (Sigma Aldrich), an oil soluble base which raised the precursor pH and initiated rapid crosslinking. The emulsion was vortexed for an additional 20 s and stirred at room temperature overnight in the dark to ensure crosslinking was complete. Microparticles were centrifuged, washed in mineral oil 5 times and hexane 5 times before resuspending in 70% ethanol. Microparticles were then sieved twice against 70 μm cell strainers and stored at 4 °C in 70% ethanol. Microparticles were rinsed three times in PBS immediately before use. Microparticle diameters were manually measured using ImageJ (NIH) using at least 500 microparticles of each type across three batches to estimate size distributions.
Formation of HA-MP and PEG-MP Scaffolds
PEG-MP scaffolds were prepared by mixing a 1:3 volumetric ratio of NP-HA precursor solution to PEG microparticles. Scaffolds were formed by incubation at 37 °C for 2 h to allow time for the NP-HA hydrogel to crosslink around the PEG microparticles. After encapsulation with NP-HA hydrogels, degradability of FITC-tagged microparticles was confirmed by submerging PEG-MP scaffolds in varying concentrations of TrypLE enzyme and observing degradation of particles by confocal fluorescence microscopy. HA-MP scaffolds were formed by first vortexing the purified microparticles before rinsing in PBS and removing supernatant. HA microparticles were injected directly into 8-mm silicon isolators and incubated for 2 h at 37 °C. Crosslinking was confirmed in vitro by rinsing with PBS with or without 1 mg/mL disulfide-reducing dithiothreitol (DTT, Sigma Aldrich) (data not shown). HA-MP scaffolds were considered formed if they disassembled only in the presence of DTT.
Mechanical Characterization of Scaffolds
Scaffolds were swollen to equilibrium overnight in PBS before mechanical characterization. Rheological characterization was done using an AR2000 rheometer (TA Instruments) with crosshatched plate and 8 mm geometry to prevent slipping and a solvent trap to minimize evaporation. The testing stage was maintained at 37 °C. Oscillatory stress sweeps at 1 Hz and 1% strain and frequency sweeps at 1% strain were performed on scaffolds swollen in PBS, pH 7.4.
Confocal Microscopy to Evaluate Scaffold Macrostructure
Scaffolds were imaged using an SP5 confocal microscope (Leica Microsystems) after being tagged using Texas red maleimide (Life Technologies) for NP-HA and HA-MP scaffolds or fluorescein-5-maleimide for PEG microparticles. Texas red or fluorescein maleimide was reacted with HA-SH or thiol-containing peptide crosslinker at 0.1 mg/mL. 3D reconstructions were created using the volume viewer plugin for ImageJ (NIH). To assess pore interconnectivity, scaffolds were formed without fluorescent labels and were incubated in a 1 mg/mL solution of high molecular weight (500 kDa) fluorescein isothiocyanate (FITC)-dextran (TdB Consultancy AB). FITC-dextran was incubated with Texas-red tagged NP-HA scaffolds to confirm the inability to diffuse into nanoporous scaffolds (Fig. S1A). Void space was calculated by thresholding images using an Otsu algorithm and calculating the percent area covered by FITC-dextran throughout the scaffold volume.
Hydraulic Conductivity to Assess Scaffold Macrostructures
Macroporous structures of scaffolds were further assessed through measurements of hydraulic conductivity. A 3D-printed device was used to fix scaffolds between a 5 μm pore size cellulose membrane on the bottom and a 500 µL chamber of PBS on top (Fig. S1B). The rate of flow of PBS through the scaffold was measured over at least 3 h and used to calculate hydraulic conductivity by the falling-head method wherein buffer is not replenished and pressure through the system begins at the same point but falls over time.1 Hydraulic conductivity (K) is derived from Darcy’s Law as L/Δt*ln(hf/hi), where L is the scaffold thickness, hf is the final buffer height, and hi is the initial buffer height.
In Vivo Delivery of Gene-Loaded Biomaterial Scaffolds
All in vivo studies were conducted in compliance with the NIH Guide for Care and Use of Laboratory Animals and with approval from the UCLA Institutional Animal Care and Use Committee. Studies were performed on 8–10 weeks old healthy female C57BL mice (Charles River) with ad libitum access to food and water. Mice were anesthetized using vaporized isoflurane and mouse mammary fat pads were exposed. 15 μL of PEG-MP or HA-MP scaffold precursor was injected into the left or right fat pad, with the opposite side acting as an NP-HA internal control. Lentivirus was mixed with scaffold precursor (108–109 particles/scaffold) immediately prior to injection. In all cases, 100 µg/mL of poly-L-lysine (30-70kDa, Sigma Aldrich) was added to precursor solutions to promote viral stability and cell adhesion. The incision was sealed using wound clips (9mm, BD Biosciences). When delivering FLuc-LV, in vivo luciferase imaging was performed at 4- and 8-days post-injection. Mice (n = 4–6) were euthanized after 9 days. Scaffolds were extracted, fixed, and cryosectioned for histology and immunohistochemistry. Additional mice were injected with HA-MP scaffolds containing td-tomato-expressing lentivirus (n = 2, 107–108 particles/scaffold) to enable identification of transduced cells. Scaffolds were fixed in 4% paraformaldehyde (PFA) overnight at 4 °C and frozen in optimal cutting temperature (OCT) compound before cryosectioning (18 μm thick). Cryosectioning and hematoxylin & eosin (H&E) staining were performed by the Translational Pathology Core Laboratory at UCLA.
Immunofluorescence Staining and Analysis
Tissue cryosections were post-fixed in 4% PFA for 12 min, blocked for 1 h with 10% normal donkey or goat serum, depending on the secondary antibody host, and 5% bovine serum albumin, incubated overnight with primary antibodies at 4 °C, secondary antibodies for 45 min at room temperature, and nuclei stain for 3 min. Sections were washed with tris-buffered saline with 0.1% tween-20 (TBST) between steps. Coverslips were mounted onto stained sections using Fluoromount G mounting medium (Southern Biotech). Primary antibodies used were rat anti-F4/80 (1:200, MCA497R, AbD Serotec) to detect macrophages, rat anti-ERTR7 (1:200, sc-73355, Santa Cruz Biotechnology) to detect fibroblasts, rat anti-CD31 (1:200, BDB553370, BD Biosciences) to detect endothelial cells, and goat anti-td tomato (1:200, LS-C340696, LifeSpan BioSciences, Inc.) to detect transduced cells. Hoechst 33342 (1:2000, H3570, Life Technologies) counterstain was used to identify nuclei. Secondary antibodies against rat (1:1000, SAB4600133, donkey anti-rat, Sigma-Aldrich) or goat (1:1000, SAB4600032, donkey anti-goat, Sigma-Aldrich) were used, as appropriate. Wide-field fluorescence images were taken using an Axio Observer microscope (Carl Zeiss) at × 200 magnification with numerical aperture of 0.8. Staining and imaging were performed in a single batch and using identical exposure levels for direct comparison. Images were analyzed using custom MATLAB software with the Image Processing toolbox and CellProfiler software.30 Cell densities at varying distances from the scaffold border were approximated based on area covered by nuclei staining of thresholded images using custom MATLAB code. For each animal, five cryosections were stained and analyzed, covering at least 90 µm of scaffold thickness. Integrated intensity per area within scaffolds was calculated separately for images of F4/80, ERTR7, and CD31 stained sections. HA-MP scaffolds were further assessed for overlapping td-tomato + and cell marker + (i.e., F4/80, ERTR7, or CD31) areas within the scaffold in thresholded, stained images.
Statistical Analyses
Data were analyzed using GraphPad Prism 6 software. Differences in scaffold properties, cell counts, and immunostaining of cell types were analyzed using Kruskal–Wallis test followed by Dunn’s multiple comparisons test. Data are displayed as mean ± standard error of the mean with significance considered to be p < 0.05 (*p < 0.05, **p<0.01, ***p < 0.001, ****p < 0.0001). Nuclei staining density within scaffolds (thresholded nuclei+ area / scaffold area) was analyzed via two-way ANOVA, where independent variables were time and scaffold type. Differences in luciferase expression was analyzed using a nonparametric bootstrapping approach.55 Two-sided 95% confidence intervals were calculated with 10,000 iterations using MATLAB software loaded with the Statistics and Machine Learning Toolbox. Significance denotes no overlap between 95% confidence intervals.
Results
Both degradable porogen and annealed microparticles produced macroporous scaffolds (PEG-MP and HA-MP, respectively) in vitro and were compatible with injection and in situ formation within the mouse fat pad. Macroporous and nanoporous scaffolds were developed from the same polymer backbones, high molecular weight HA and PEG, to facilitate direct comparisons of the effects of porous architecture, as opposed to scaffold chemistry, on tissue integration and transgene delivery. Material properties of scaffolds, including void fraction, hydraulic conductivity and mechanical moduli, were first characterized in vitro. After mixing HA-SH and PEG-VS precursor components, crosslinking occurred within 30 min at 37 °C but handled like a liquid for over 2 h at room temperature. This reaction rate enabled precursor solutions to be mixed prior to injection in vivo, where crosslinking could then occur in situ to form both NP-HA and PEG-MP scaffolds. HA-MP scaffolds could be similarly handled at room temperature and formed in situ. Microparticles within PEG-MP scaffolds degraded within 24 h in vitro when exposed to proteolytic enzymes at 10 ng/mL or greater (Table S1). Microparticles composing PEG-MP and HA-MP scaffolds had diameters of 20 ± 9 µm and 42 ± 23 µm, respectively (Fig. S2). Rheological measurements of shear elastic moduli of the formed NP-HA and HA-MP scaffolds were comparable at 160 ± 32 Pa and 100 ± 20 Pa, respectively, but PEG-MP scaffolds had significantly greater moduli at 405 ± 30 Pa prior to particle degradation (Fig. S3). It is important to note that PEG-MP scaffolds had greater moduli as a direct result of degradable PEG particles, which were intact (i.e., not yet degraded) for these measurements. As PEG particles were encapsulated in hydrogels formulated identically to NP-HA scaffolds. Therefore, we expect the microscale modulus that cells encountering scaffolds, after degradation of PEG particle porogens, would experience to be comparable to that of NP-HA scaffolds.
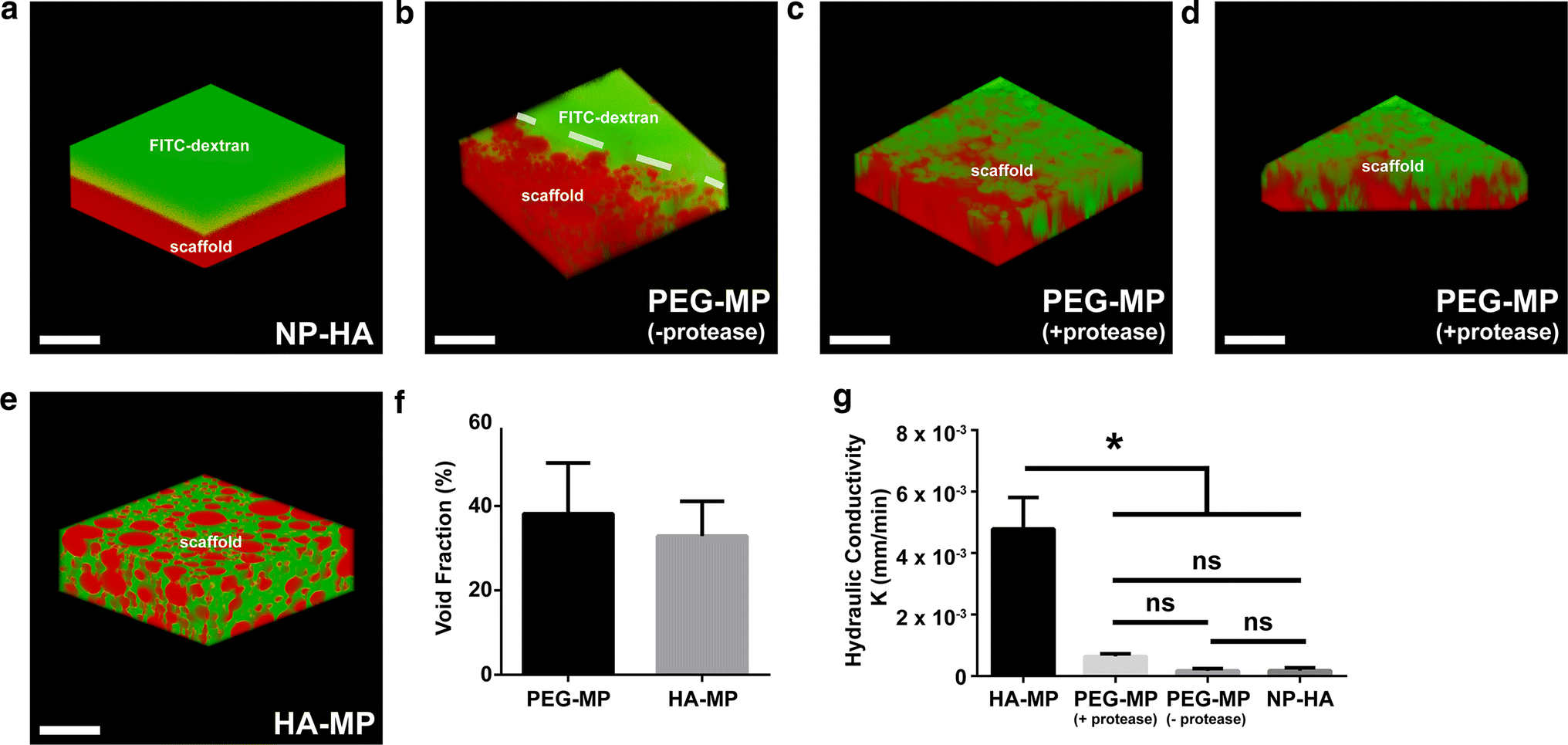
Characterization of microporous networks in PEG-MP and HA-MP scaffolds. Confocal reconstructions of scaffolds after incubation with high molecular weight (Mw = 500 kDa) FITC-dextran indicates interconnectivity of a macroscale void space through PEG-MP and HA-MP, but not NP-HA, scaffolds (a–e). In all cases, FITC-Dextran is represented in green. NP-HA scaffolds and microparticles in PEG-MP scaffolds were tagged by Texas red-maleimide. For PEG-MP scaffolds in which microparticle porogens have already been degraded and in HA-MP scaffolds, an inverse signal of FITC-dextran (displayed in red) is used to accentuate the key features of clear spherical HA-MP particles or pockets in PEG-MP scaffolds. Reconstructions show no void space in NP-HA scaffolds (a), an inability for FITC-dextran to diffuse into the edge of PEG-MP scaffolds before particle degradation (b), inconsistent infiltration in PEG-MP scaffolds over 200 µm from the scaffold surface (c) which decreased towards the scaffold center (bottom of reconstructed 3D image shown) (d), and consistent infiltration through the void space in HA-MP scaffolds (e). Quantification shows roughly 35–40% void fraction in both PEG-MP and HA-MP scaffolds over a 200 µm depth from scaffold surfaces (f). Hydraulic conductivity based on the rate at which buffer travels through the scaffolds shows the greatest open interconnectivity of macropores in HA-MP scaffolds and the least in NP-HA scaffolds (g). Error bars represent standard deviation (*p < 0.05, Kruskal–Wallis test with Dunn’s post-hoc tests, n = 4). Scale bars = 200 µm.

Transgene expression increased when lentiviral vectors were delivered from macroporous scaffolds. Expression of a bioluminescent transgene, firefly luciferase (FLuc), in HA-MP and PEG-MP scaffolds was 3.6-fold and 1.3-fold greater (on average), respectively, than in NP-HA scaffolds at day 8. Scaffolds were initially loaded with 2.0 × 108 active lentiviral particles per NP-HA or HA-MP scaffold, and 1.2 × 108 per PEG-MP scaffold (a, b). Increasing the initial viral load by approximately 6.5-fold resulted in a proportional increase in transgene expression around HA-MP scaffolds, but NP-HA and PEG-MP scaffolds were not significantly affected. Mean average radiance from HA-MP scaffolds was as much as 16-fold greater than that of NP-HA or PEG-MP scaffolds by day 8 (c, d). (*p < 0.05, nonparametric bootstrapping with 10,000 iterations, n = 7–9, mean of the data set is denoted by a horizontal line).

Host cell infiltration into NP-HA, PEG-MP and HA-MP scaffolds explanted after 9 days. H&E staining (a) and Hoechst nuclear counterstain (b) near the host-scaffold interface. Densities of cell nuclei were significantly greater at the center of HA-MP than in NP-HA or PEG-MP scaffolds (p < 0.05, n = 7, Kruskal–Wallis test) (c). Quantification of densities of cell nuclei across scaffolds from the tissue interface to the center demonstrated greater penetration depth into HA-MP over other scaffolds (*p < 0.05, n = 7–9, two-way ANOVA, Tukey’s post-hoc tests) (d). White dashed lines indicate the interfaces between scaffolds and host tissues. Scaffolds were initially loaded with 1.3 × 109 active lentiviral particles per NP-HA or HA-MP scaffold, and 9.8 × 108 per PEG-MP scaffold. Scaffolds are indicated on the left-hand side of the dashed lines. Scale bars = 200 µm.

Macrophages, fibroblasts and endothelial cells are found to infiltrate scaffolds. F4/80+ cells were present throughout NP-HA, PEG-MP, and HA-MP scaffolds (a). F4/80+ intensity/area was statistically equivalent for all scaffolds (b). ERTR7+ cells were also present within all scaffolds (c) with significantly less intensity/area in NP-HA than in HA-MP scaffolds (d). PEG-MP scaffolds had visually greater immunostaining and intensity/area for ERTR7 than NP-HA scaffolds but were not significantly different than NP-HA or HA-MP scaffolds. CD31+ cells were most notably present in HA-MP scaffolds, with some presence in PEG-MP scaffolds (e). Significantly greater CD31+ intensity/area was observed in HA-MP compared to NP-HA, but not PEG-MP, scaffolds (*p < 0.05, Kruskal–Wallis test with Dunn’s post-hoc tests, n = 4–5) (f). CD31+ staining is clear at depths of over 200 µm in HA-MP but not PEG-MP or NP-HA scaffolds (G). Scaffolds were initially loaded with 1.3 × 109 active lentiviral particles per NP-HA or HA-MP scaffold, and 9.8 × 108 per PEG-MP scaffold. Scaffolds are indicated on the left-hand side of the dashed lines. Scale bars = 200 µm, 20 µm for zoomed inset. Arrows indicate F4/80, ERTR7, or CD31 positive cells within scaffolds.
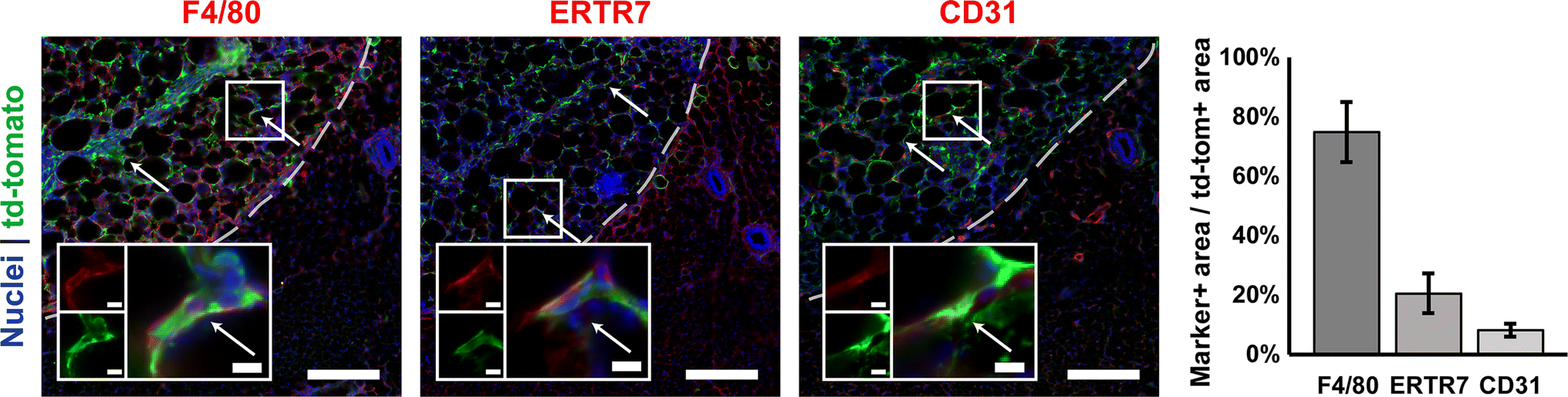
Cells expressing the td-tomato transgene are located within scaffolds. Td-tomato expression overlapped with F4/80+, ERTR7+, and CD31+ positive areas, in order of with decreasing abundance. Arrows indicate F4/80, ERTR7, or CD31 positive cells which express td-tomato. Scaffolds were initially loaded with 3.5 × 108 active lentiviral particles. Scaffolds are located on the left-hand side of the dashed lines. Zoomed insets show examples of double-positive cells (RGB overlays and separate red and green channels). Scale bars = 200 µm, 10 µm for zoomed insets (n = 2).
Discussion
Injectable scaffolds can provide efficient, minimally invasive, and highly localized delivery of therapeutic transgenes. Previous studies have demonstrated that the efficiency of scaffold-mediated transgene delivery depends on robust and rapid infiltration of host cells, which can then physically encounter vectors and uptake transgenes. Early infiltration of host cells into biomaterial scaffolds has been maximized through incorporation of cell-scale, macroporous networks.43,50 While various strategies have been developed in recent years for fabrication of scaffolds that are injectable and macroporous,4,18,19,52,53 the relative abilities of these scaffolds to deliver transgene in vivo has not yet been investigated. Here, data clearly demonstrate that injection of lentiviral-loaded, HA-based microparticles and in situ assembly into macroporous scaffolds (i.e., HA-MPs) significantly increases host cell expression of delivered transgenes. In comparison, injectable PEG-MP scaffolds, in which macropore generation requires migrating cells to proteolytically degrade PEG microparticles, only showed modest improvements in transgene expression, compared to nanoporous, NP-HA scaffolds.
NP-HA, PEG-MP, and HA-MP scaffolds were each able to be injected into the soft tissue of mouse mammary fat pads and subsequently crosslink in situ to form scaffolds that tightly interfaced with host tissues. To deliver lentiviral vectors from scaffolds, they must be added to precursor solutions immediately prior to injection, as third generation lentiviral particles degrade quickly in vivo, with a half-life reported to be as low as 12 h at 37 °C.21,48 Dilution with lentiviral solution necessitates that scaffold precursors be prepared at higher densities than are desired for injection. Thus, the viscosity and solubility limits of these precursor solutions restrict the maximum dose of lentivirus that can be loaded. Furthermore, dilution likely affects the final architecture of macropores in PEG-MP scaffolds, as high densities of degradable microparticles suspended in hydrogel precursor solution prior to injection are required to create an interconnected network through which cells can migrate after in situ crosslinking.
FLuc was used as a model transgene, enabling tracking of its expression in vivo over time via bioluminescence imaging. In all scaffolds, FLuc expression was highly localized to the site of injection. HA-MP scaffolds most efficiently promoted transgene expression and were the only scaffolds in which FLuc expression increased from days 4 to 8 after implantation. While a higher proliferation rate of transduced cells within HA-MP scaffolds is a possible explanation for this finding, given the greater extent of cell infiltration into HA-MP scaffolds as seen in histological analysis, it is more likely that higher numbers of cells were initially transduced and that proliferation of these cells simply amplified relative differences in transgene expression over time.
While PEG-MP scaffolds showed slightly greater FLuc expression than NP-HA scaffolds in many cases, these differences were not significant. Microparticles within PEG-MP scaffolds degraded within 24 h when exposed to physiological concentrations of proteolytic enzymes49in vitro (Table S1). Additionally, cells in 3D culture within hydrogels with identical chemistry (PEG-VS crosslinked with the same fast-degrading, plasmin-susceptible peptide) have been reported to degrade in under 4 days.44 Given these findings, we expect that PEG microparticles within PEG-MP scaffolds would have degraded during this time-frame in vivo. Thus, it is more likely that microparticle density was not high enough to achieve a truly interconnected macroporous network after degradation, which would prevent infiltration of cells throughout the scaffolds, resulting in relatively lower transduction efficiencies.47 Comparisons of hydraulic conductivity measurements, which were significantly higher for HA-MP than for PEG-MP or NP-HA scaffolds, support this idea. Similarly, NP-HA scaffolds limit transduction of host cells because they cannot infiltrate scaffolds and contact lentivirus without first degrading the crosslinked HA matrix, which typically takes weeks in vivo.2 While it is likely possible to achieve higher interconnectivity of pores generated in scaffolds using from degradable microparticles as templates, the high packing densities, typically 50–80%,22,23,47 that are likely required to achieve this impose restrictions on the scaffold design that may preclude their use for some applications. In the scaffolds used here, higher packing density of microparticle porogens would alter bulk material properties and reduce exposure of host tissue to the bioactive benefits of HA for wound healing. Thus, this method may be most appropriate for applications where targeted degradation of microparticle porogens (e.g., by a particular enzyme or other biological trigger) is required, and not for more general applications of scaffold-mediated gene delivery where the main determinant of transduction is rapid formation of pores followed by maximal cell infiltration.
In general, the extent of host cell infiltration into scaffolds reflected the efficiency of transgene expression. HA-MP scaffolds, which yielded the greatest transgene expression (as high as 16-fold increase in mean bioluminescence average radiance over that in NP-HA scaffolds), also had the greatest cell densities within scaffold centers (around 8.1-fold more cells than in NP-HA scaffolds). Cell densities at the center of PEG-MP scaffolds were slightly greater than NP-HA scaffolds (by approximately 3.6-fold), though this effect was not statistically significant. Cell density was analyzed using an automated program that occasionally identified clusters of individual cells as single cells, especially in areas of high cell density, so that total numbers of cells were likely underestimated. Therefore, the actual cell density in HA-MP scaffolds was likely even greater than our quantification indicated. Unlike NP-HA and PEG-MP scaffolds, cell migration through HA-MP scaffolds does not depend on degradation of any hydrogel network. Overall, these results indicate that the immediate presence of an interconnected, macroporous network after injection can increase the number of cells that are able to infiltrate scaffolds prior to lentiviral vector degradation and thus dramatically increase levels of transgene expression.
Despite no clear differences in cell infiltration when more lentiviral particles were loaded into scaffolds, HA-MP scaffolds still showed a significant increase in transgene expression. This indicates that either more infiltrating cells were able to be transduced or that similar numbers of cells were transduced, but by multiple lentiviral vectors. While cells transduced with more copies of a transgene will generally have higher expression levels, there is also a higher chance that transgene insertion will occur at points in the genome that disrupt cell function or activate oncogenes. Furthermore, increasing the concentration of lentivirus delivered did not have any observable effects on fibrotic or immune cell responses (Fig. S6B, C).
Explanted sections were immunostained to identify macrophages (F4/80+), fibroblasts (ERTR7+), and endothelial cells (CD31+) that had infiltrated scaffolds. All scaffolds were abundant in F4/80+ macrophages, which would be expected near foreign objects, such as biomaterial implants. However, analysis of integrated intensity of immunofluorescence per area showed substantially more ERTR7 and CD31 staining in HA-MP than NP-HA scaffolds. While average integrated intensities for ERTR7 and CD31 in PEG-MP scaffolds generally fell between those for NP-HA and HA-MP scaffolds, there were no statistically significant differences. The macroscale pores in HA-MP and PEG-MP scaffolds may allow for greater infiltration of non-immune cells, such as fibroblasts and endothelial cells. CD31 immunostaining indicated signs of angiogenesis, with vessel-like structures present that appeared similar to those in prior reports where scaffolds were either seeded with endothelial cells in vitro or engineered to induce angiogenesis in vivo.13,37,51,58 However, further studies will need to be performed to definitively assess the extent to which angiogenesis occurs within these scaffolds. Numerous studies have emphasized the importance of scaffold vascularization for wound healing and tissue engineering applications to facilitate oxygen, nutrient, and waste transport to cells present throughout scaffolds.28 Furthermore, scaffolds supporting early vasculogenesis have increased potential to improve wound healing.2,3,18 While CD31 is a fairly specific marker for endothelial cells and staining was mostly confined to blood vessel-like structures and F4/80 has been found to distinguish macrophages from adipocytes and fibroblasts in mice.7,27 While ERTR7 have been reported to be expressed in fat-storing cells in the mouse thymus,10 other studies reported that ERTR7 sufficiently distinguished fibroblasts from adipocytes in mouse dermal40 and immune14 (e.g., spleen and bone marrow) tissues. In the current study, many of the ERTR7+ cells outside of scaffolds did appear to exhibit more adipocyte-like, rather than fibroblast-like, morphologies. However, we suspect that ERTR7+ cells within scaffolds were fibroblasts, and not adipocytes, given their morphology (Figs. 5c and 6) and that adipocytes are not generally migratory.
Given the short half-life of lentiviral particles,34 it is more likely that highly migrating cells, including macrophages and fibroblasts would be transduced. In contrast, cells entering scaffolds after lentiviral activity has degraded would not uptake vectors or express transgene. In HA-MP scaffolds, co-immunostaining for the delivered td-tomato transgene and cell-type specific markers indicated that transgene expressing cells were either macrophages (F4/80+), fibroblasts (ERTR7+), or endothelial cells (CD31+). The majority of td-tomato-expressing cells appeared to F4/80+ macrophages, with some fibroblasts and only a few endothelial cells, indicating that macrophages infiltrate scaffolds in large numbers very quickly after injection. In line with bioluminescence imaging, transgene expression was largely confined to cells within the scaffolds, demonstrating the utility of such injectable, macroporous scaffolds for applications where highly localized transgene delivery is required; for example, to avoid negative side effects possibly associated with systemic delivery. In line with previous reports in vitro, this result indicates that addition of poly-l-lysine into scaffolds was sufficient to retain active lentivirus and confine transduction to scaffolds in vivo.46
Conclusion
Biomaterial scaffolds with cell-scale, macroporous architectures can improve early infiltration of host cells after implantation to enhance uptake of immobilized genetic vectors. Here, we compared the gene delivery efficiencies of two strategies for creating macroporous scaffolds that can be injected directly into tissue and formed in situ. We expect that this capability will be useful for a number of different applications in regenerative medicine where localized delivery of gene therapies to irregularly shaped or difficult to access sites is required. Results demonstrate that transduction by lentiviral particles loaded into biomaterials was maximized when scaffolds with an open, macroporous architecture were created through in situ assembly of pre-crosslinked, hydrogel microparticles. This strategy eliminated the need for cells to degrade scaffold materials during infiltration and supported rapid migration of cells, particularly macrophages, throughout scaffolds, maximizing the number of these cells that became transduced prior to a loss of lentiviral activity.
Notes
Acknowledgments
The authors would like to acknowledge funding for this work from a National Science Foundation CAREER Award 1653730 (SKS), a UCLA Henry Samueli School of Engineering and Applied Sciences (HSSEAS) Faculty Research Grant (SKS) and a UCLA Faculty Career Development Award (SKS). We thank the UCLA Tissue Pathology Core Laboratory (TPCL) for cryosectioning and hematoxylin and eosin staining, the UCLA Crump Institute for Molecular Imaging for use of the IVIS imaging system, and the UCLA Molecular Instrumentation Center for use of proton NMR facilities. Confocal laser scanning microscopy was performed at the California NanoSystems Institute Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA, supported with funding from NIH-NCRR shared resources grant (CJX1-443835-WS-29646) and NSF Major Research Instrumentation Grant (CHE-0722519).
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical Standards
All animal studies were carried out in accordance with the NIH Guide for Care and Use of Laboratory Animals and approved by the UCLA Institutional Animal Care and Use Committee. No human studies were carried out by the authors for this article.
Supplementary material
References
-
1.Angulo-Jaramillo, R., J.-P. Vandervaere, S. Roulier, J.-L. Thony, J.-P. Gaudet, and M. Vauclin. Field measurement of soil surface hydraulic properties by disc and ring infiltrometers: A review and recent developments. Soil Tillage Res. 55:1–29, 2000.CrossRefGoogle Scholar
-
2.Baier Leach, J., K. A. Bivens, C. W. Patrick, Jr, and C. E. Schmidt. Photocrosslinked hyaluronic acid hydrogels: natural, biodegradable tissue engineering scaffolds. Biotechnol. Bioeng. 82:578–589, 2003.CrossRefGoogle Scholar
-
3.Baranski, J. D., et al. Geometric control of vascular networks to enhance engineered tissue integration and function. Proc. Natl. Acad. Sci. 110:7586–7591, 2013.CrossRefGoogle Scholar
-
4.Bencherif, S. A., et al. Injectable preformed scaffolds with shape-memory properties. Proc. Natl. Acad. Sci. USA 109:19590–19595, 2012.CrossRefGoogle Scholar
-
5.Bernabé, B. P., S. Shin, P. D. Rios, L. J. Broadbelt, L. D. Shea, and S. K. Seidlits. Dynamic transcription factor activity networks in response to independently altered mechanical and adhesive microenvironmental cues. Integr. Biol. 8:844–860, 2016.CrossRefGoogle Scholar
-
6.Caldwell, A. S., G. T. Campbell, K. M. T. Shekiro, and K. S. Anseth. Clickable microgel scaffolds as platforms for 3D cell encapsulation. Adv. Healthc. Mater. 6:1700254, 2017.CrossRefGoogle Scholar
-
7.Cao, Q., et al. Renal F4/80+CD11c+ mononuclear phagocytes display phenotypic and functional characteristics of macrophages in health and in adriamycin nephropathy. J. Am. Soc. Nephrol. 26:349–363, 2015.CrossRefGoogle Scholar
-
8.Chau, Y., Y. Yu, L. Lau, and A. C. Lo. In vivo evaluation of hyaluronic acid based in situ hydrogel for prolonged release of Avastin by intravitreal injection. Invest. Ophthalmol. Vis. Sci. 55:5261, 2014.Google Scholar
-
9.Chiu, Y.-C., J. C. Larson, A. Isom, and E. M. Brey. Generation of porous poly(ethylene glycol) hydrogels by salt leaching. Tissue Eng. C 16:905–912, 2010.CrossRefGoogle Scholar
-
10.de Mello Coelho, V., et al. Fat-storing multilocular cells expressing CCR1 increase in the thymus with advancing age: potential role for CCR1 ligands on the differentiation and migration of preadipocytes. Int. J. Med. Sci. 7:1–14, 2009.Google Scholar
-
11.Dull, T., et al. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471, 1998.Google Scholar
-
12.Ellman, G. L. A colorimetric method for determining low concentrations of mercaptans. Arch. Biochem. Biophys. 74:443–450, 1958.CrossRefGoogle Scholar
-
13.Ferreira, L. S., S. Gerecht, J. Fuller, H. F. Shieh, G. Vunjak-Novakovic, and R. Langer. Bioactive hydrogel scaffolds for controllable vascular differentiation of human embryonic stem cells. Biomaterials 28:2706–2717, 2007.CrossRefGoogle Scholar
-
14.Gil-Ortega, M., et al. Native adipose stromal cells egress from adipose tissue in vivo: evidence during lymph node activation. Stem Cells Dayt. Ohio 31:1309–1320, 2013.CrossRefGoogle Scholar
-
15.Gong, P., G. M. Harbers, and D. W. Grainger. Multi-technique comparison of immobilized and hybridized oligonucleotide surface density on commercial amine-reactive microarray slides. Anal. Chem. 78:2342–2351, 2006.CrossRefGoogle Scholar
-
16.Gong, Y., Z. Ma, Q. Zhou, J. Li, C. Gao, and J. Shen. Poly(lactic acid) scaffold fabricated by gelatin particle leaching has good biocompatibility for chondrogenesis. J. Biomater. Sci. Polym. Ed. 19:207–221, 2008.CrossRefGoogle Scholar
-
17.Gonzalez-Fernandez, T., E. G. Tierney, G. M. Cunniffe, F. J. O’Brien, and D. J. Kelly. Gene delivery of TGF-β3 and BMP2 in an MSC-laden alginate hydrogel for articular cartilage and endochondral bone tissue engineering. Tissue Eng. A 22:776–787, 2016.CrossRefGoogle Scholar
-
18.Griffin, D. R., W. M. Weaver, P. O. Scumpia, D. Di Carlo, and T. Segura. Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks. Nat. Mater. 14:737–744, 2015.CrossRefGoogle Scholar
-
19.Han, L.-H., S. Yu, T. Wang, A. W. Behn, and F. Yang. Microribbon-like elastomers for fabricating macroporous and highly flexible scaffolds that support cell proliferation in 3D. Adv. Funct. Mater. 23:346–358, 2013.CrossRefGoogle Scholar
-
20.Hiemstra, C., L. J. van der Aa, Z. Zhong, P. J. Dijkstra, and J. Feijen. Rapidly in situ-forming degradable hydrogels from dextran thiols through Michael addition. Biomacromolecules 8:1548–1556, 2007.CrossRefGoogle Scholar
-
21.Higashikawa, F., and L.-J. Chang. Kinetic analyses of stability of simple and complex retroviral vectors. Virology 280:124–131, 2001.CrossRefGoogle Scholar
-
22.Huebsch, N., et al. Matrix elasticity of void-forming hydrogels controls transplanted-stem-cell-mediated bone formation. Nat. Mater. 14:1269–1277, 2015.CrossRefGoogle Scholar
-
23.Hwang, C. M., et al. Fabrication of three-dimensional porous cell-laden hydrogel for tissue engineering. Biofabrication 2:035003, 2010.CrossRefGoogle Scholar
-
24.Ibrahim, S., Q. K. Kang, and A. Ramamurthi. The impact of hyaluronic acid oligomer content on physical, mechanical, and biologic properties of divinyl sulfone-crosslinked hyaluronic acid hydrogels. J. Biomed. Mater. Res. A 94A:355–370, 2010.Google Scholar
-
25.Jain, A., Y.-T. Kim, R. J. McKeon, and R. V. Bellamkonda. In situ gelling hydrogels for conformal repair of spinal cord defects, and local delivery of BDNF after spinal cord injury. Biomaterials 27:497–504, 2006.CrossRefGoogle Scholar
-
26.Johnson, P. J., A. Tatara, A. Shiu, and S. E. Sakiyama-Elbert. Controlled release of neurotrophin-3 and platelet-derived growth factor from fibrin scaffolds containing neural progenitor cells enhances survival and differentiation into neurons in a subacute model of SCI. Cell Transplant. 19:89–101, 2010.CrossRefGoogle Scholar
-
27.Khazen, W., et al. Expression of macrophage-selective markers in human and rodent adipocytes. FEBS Lett. 579:5631–5634, 2005.CrossRefGoogle Scholar
-
28.Kim, J. J., L. Hou, and N. F. Huang. Vascularization of three-dimensional engineered tissues for regenerative medicine applications. Acta Biomater. 41:17–26, 2016.CrossRefGoogle Scholar
-
29.Kong, H. J., E. S. Kim, Y.-C. Huang, and D. J. Mooney. Design of biodegradable hydrogel for the local and sustained delivery of angiogenic plasmid DNA. Pharm. Res. 25:1230–1238, 2008.CrossRefGoogle Scholar
-
30.Lamprecht, M. R., D. M. Sabatini, and A. E. Carpenter. Cell Profiler: free, versatile software for automated biological image analysis. BioTechniques 42:71–75, 2007.CrossRefGoogle Scholar
-
31.Litwiniuk, M., A. Krejner, M. S. Speyrer, A. R. Gauto, and T. Grzela. Hyaluronic acid in inflammation and tissue regeneration. Wounds Compend. Clin. Res. Pract. 28:78–88, 2016.Google Scholar
-
32.Liu, S., et al. Regulated viral BDNF delivery in combination with Schwann cells promotes axonal regeneration through capillary alginate hydrogels after spinal cord injury. Acta Biomater. 60:167–180, 2017.CrossRefGoogle Scholar
-
33.Margul, D. J., et al. Reducing neuroinflammation by delivery of IL-10 encoding lentivirus from multiple-channel bridges. Bioeng. Transl. Med. 1:136–148, 2016.CrossRefGoogle Scholar
-
34.Nikolaev, S. I., A. R. Gallyamov, G. V. Mamin, and Y. A. Chelyshev. Poly(ε-caprolactone) nerve conduit and local delivery of vegf and fgf2 genes stimulate neuroregeneration. Bull. Exp. Biol. Med. 157:155–158, 2014.CrossRefGoogle Scholar
-
35.Patel, Z. S., S. Young, Y. Tabata, J. A. Jansen, M. E. K. Wong, and A. G. Mikos. Dual delivery of an angiogenic and an osteogenic growth factor for bone regeneration in a critical size defect model. Bone 43:931–940, 2008.CrossRefGoogle Scholar
-
36.Perumcherry, S. R., K. P. Chennazhi, S. V. Nair, D. Menon, and R. Afeesh. A novel method for the fabrication of fibrin-based electrospun nanofibrous scaffold for tissue-engineering applications. Tissue Eng. C 17:1121–1130, 2011.CrossRefGoogle Scholar
-
37.Peterson, A. W., D. J. Caldwell, A. Y. Rioja, R. R. Rao, A. J. Putnam, and J. P. Stegemann. Vasculogenesis and angiogenesis in modular collagen-fibrin microtissues. Biomater. Sci. 2:1497–1508, 2014.CrossRefGoogle Scholar
-
38.Poursamar, S. A., J. Hatami, A. N. Lehner, C. L. da Silva, F. C. Ferreira, and A. P. M. Antunes. Gelatin porous scaffolds fabricated using a modified gas foaming technique: characterisation and cytotoxicity assessment. Mater. Sci. Eng. C 48:63–70, 2015.CrossRefGoogle Scholar
-
39.Saraf, A., L. S. Baggett, R. M. Raphael, F. K. Kasper, and A. G. Mikos. Regulated non-viral gene delivery from coaxial electrospun fiber mesh scaffolds. J. Control. Release 143:95–103, 2010.CrossRefGoogle Scholar
-
40.Schmidt, B. A., and V. Horsley. Intradermal adipocytes mediate fibroblast recruitment during skin wound healing. Dev. Camb. Engl. 140:1517–1527, 2013.Google Scholar
-
41.Schulte, V. A., D. F. Alves, P. P. Dalton, M. Moeller, M. C. Lensen, and P. Mela. Microengineered PEG hydrogels: 3D scaffolds for guided cell growth. Macromol. Biosci. 13:562–572, 2013.CrossRefGoogle Scholar
-
42.Sheikhi, A., et al. Microfluidic-enabled bottom-up hydrogels from annealable naturally-derived protein microbeads. Biomaterials 192:560–568, 2019.CrossRefGoogle Scholar
-
43.Shepard, J. A., F. R. Virani, A. G. Goodman, T. D. Gossett, S. Shin, and L. D. Shea. Hydrogel macroporosity and the prolongation of transgene expression and the enhancement of angiogenesis. Biomaterials 33:7412–7421, 2012.CrossRefGoogle Scholar
-
44.Shikanov, A., R. M. Smith, M. Xu, T. K. Woodruff, and L. D. Shea. Hydrogel network design using multifunctional macromers to coordinate tissue maturation in ovarian follicle culture. Biomaterials 32:2524–2531, 2011.CrossRefGoogle Scholar
-
45.Sideris, E., et al. particle hydrogels based on hyaluronic acid building blocks. ACS Biomater. Sci. Eng. 2:2034–2041, 2016.CrossRefGoogle Scholar
-
46.Skoumal, M., S. Seidlits, S. Shin, and L. Shea. Localized lentivirus delivery via peptide interactions. Biotechnol. Bioeng. 113:2033–2040, 2016.CrossRefGoogle Scholar
-
47.Sokic, S., M. Christenson, J. Larson, and G. Papavasiliou. In situ generation of cell-laden porous MMP-sensitive PEGDA hydrogels by gelatin leaching. Macromol. Biosci. 14:731–739, 2014.CrossRefGoogle Scholar
-
48.Thomas, A. M., and L. D. Shea. Polysaccharide-modified scaffolds for controlled lentivirus delivery in vitro and after spinal cord injury. J. Control. Release 170:421–429, 2013.CrossRefGoogle Scholar
-
49.Thrailkill, K., G. Cockrell, P. Simpson, C. Moreau, J. Fowlkes, and R. C. Bunn. Physiological matrix metalloproteinase (MMP) concentrations: comparison of serum and plasma specimens. Clin. Chem. Lab. Med. CCLM FESCC 44:503–504, 2006.Google Scholar
-
50.Tokatlian, T., C. Cam, and T. Segura. Non-viral DNA delivery from porous hyaluronic acid hydrogels in mice. Biomaterials 35:825–835, 2014.CrossRefGoogle Scholar
-
51.Tremblay, P.-L., V. Hudon, F. Berthod, L. Germain, and F. A. Auger. Inosculation of tissue-engineered capillaries with the host’s vasculature in a reconstructed skin transplanted on mice. Am. J. Transplant. 5:1002–1010, 2005.CrossRefGoogle Scholar
-
52.Truong, N. F., et al. Microporous annealed particle hydrogel stiffness, void space size, and adhesion properties impact cell proliferation, cell spreading, and gene transfer. Acta Biomater. 94:160–172, 2019.CrossRefGoogle Scholar
-
53.Truong, N. F., S. C. Lesher-Pérez, E. Kurt, and T. Segura. Pathways governing polyethylenimine polyplex transfection in microporous annealed particle scaffolds. Bioconjug. Chem. 30:476–486, 2019.CrossRefGoogle Scholar
-
54.Wang, L., J. Shansky, C. Borselli, D. Mooney, and H. Vandenburgh. Design and fabrication of a biodegradable, covalently crosslinked shape-memory alginate scaffold for cell and growth factor delivery. Tissue Eng. A 18:2000–2007, 2012.CrossRefGoogle Scholar
-
55.Wood, M. Simple methods for estimating confidence levels, or tentative probabilities, for hypotheses instead of P values, 2017 [cited 2019 Feb 12]. http://arxiv.org/abs/1702.03129.
-
56.Xiao, W., et al. Brain-mimetic 3D culture platforms allow investigation of cooperative effects of extracellular matrix features on therapeutic resistance in glioblastoma. Cancer Res. 78:1358–1370, 2017.CrossRefGoogle Scholar
-
57.Yang, C.-Y., et al. Biocompatibility of amphiphilic diblock copolypeptide hydrogels in the central nervous system. Biomaterials 30:2881–2898, 2009.CrossRefGoogle Scholar
-
58.Zhang, W., et al. Vascularization of hollow channel-modified porous silk scaffolds with endothelial cells for tissue regeneration. Biomaterials 56:68–77, 2015.CrossRefGoogle Scholar
-
59.Zhou, Y., W. Nie, J. Zhao, and X. Yuan. Rapidly in situ forming adhesive hydrogel based on a PEG-maleimide modified polypeptide through Michael addition. J. Mater. Sci. Mater. Med. 24:2277–2286, 2013.CrossRefGoogle Scholar